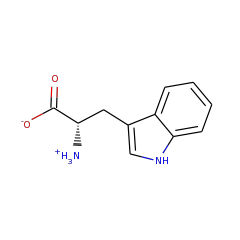
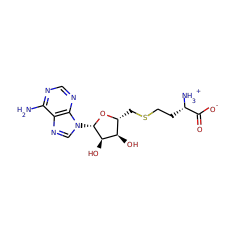
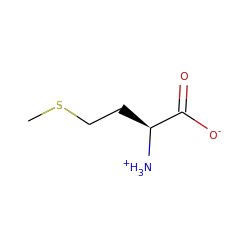
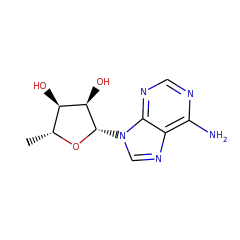
Enzymes in this family are involved in the B12 dependent methylation of aromatic carbon atoms in post-translationally modified peptides. A novel C-methylation mechanism has been proposed:
Homolysis of the cobalt–carbon bond of MeCbl by TsrM produces a methyl radical intermediate, which is transferred to the C2 position of Trp. Concomitant with Trp deprotonation, an electron is transferred to the [4Fe-4S]2+/1+ cluster, which reduces cob(II)alamin to cob(I)alamin. This supernucleophilic species can easily form MeCbl by an SN2 displacement of the methyl group of SAM-producing SAH. The double displacement of the methyl group during catalysis leads to a net configuration retention.
Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W
Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications
▸ Abstract
Thiopeptides, with potent activity against various drug-resistant pathogens, contain a characteristic macrocyclic core consisting of multiple thiazoles, dehydroamino acids, and a 6-membered nitrogen heterocycle. Their biosynthetic pathways remain elusive, in spite of great efforts by in vivo feeding experiments. Here, cloning, sequencing, and characterization of the thiostrepton and siomycin A gene clusters unveiled a biosynthetic paradigm for the thiopeptide specific core formation, featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. The paradigm generality for thiopeptide biosynthesis was supported by genome mining and ultimate confirmation of the thiocillin I production in Bacillus cereus ATCC 14579, a strain that was previously unknown as a thiopeptide producer. These findings set the stage to accelerate the discovery of thiopeptides by prediction at the genetic level and to generate structural diversity by applying combinatorial biosynthesis methods.
Chem Biol
2009;16(2):141-147
| PubMed ID:
19246004
Kelly WL, Pan L, Li C
Thiostrepton biosynthesis: prototype for a new family of bacteriocins
▸ Abstract
Thiopeptide antibiotics are a group of highly modified peptide metabolites. The defining scaffold for the thiopeptides is a macrocycle containing a dehydropiperidine or pyridine ring, dehydrated amino acids, and multiple thiazole or oxazole rings. Some members of the thiopeptides, such as thiostrepton, also contain either a quinaldic acid or indolic acid substituent derived from tryptophan. Although the amino acid precursors of these metabolites are well-established, the biogenesis of these complex peptides has remained elusive. Whole-genome scanning of Streptomyces laurentii permitted identification of a thiostrepton prepeptide, TsrA, and involvement of TsrA in thiostrepton biosynthesis was confirmed by mutagenesis. A gene cluster responsible for thiostrepton biosynthesis is reported, and the encoded gene products are discussed. The disruption of a gene encoding an amidotransferase, tsrT, led to the loss of thiostrepton production and the detection of a new metabolite, contributing further support to the identification of the tsr cluster. The tsr locus also appears to possess the gene products needed to convert tryptophan to the quinaldic acid moiety, and an aminotransferase was found to catalyze an early step in this pathway. This work establishes that the thiopeptides are a type of bacteriocin, a family of genetically encoded antimicrobial peptides, and are subjected to extensive posttranslational modification during maturation of the prepeptide.
J Am Chem Soc
2009;131(12):4327-4334
| PubMed ID:
19265401
Pierre S, Guillot A, Benjdia A, Sandström C, Langella P, Berteau O
Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes
▸ Abstract
Methylation is among the most widespread chemical modifications encountered in biomolecules and has a pivotal role in many major biological processes. In the biosynthetic pathway of the antibiotic thiostrepton A, we identified what is to our knowledge the first tryptophan methyltransferase. We show that it uses unprecedented chemistry to methylate inactivated sp(2)-hybridized carbon atoms, despite being predicted to be a radical SAM enzyme.
Nat Chem Biol
2012;8(12):957-959
| PubMed ID:
23064318
Blaszczyk AJ, Silakov A, Zhang B, Maiocco SJ, Lanz ND, Kelly WL, Elliott SJ, Krebs C, Booker SJ
Spectroscopic and Electrochemical Characterization of the Iron-Sulfur and Cobalamin Cofactors of TsrM, an Unusual Radical S-Adenosylmethionine Methylase
▸ Abstract
TsrM, an annotated radical S-adenosylmethionine (SAM) enzyme, catalyzes the methylation of carbon 2 of the indole ring of l-tryptophan. Its reaction is the first step in the biosynthesis of the unique quinaldic acid moiety of thiostrepton A, a thiopeptide antibiotic. The appended methyl group derives from SAM; however, the enzyme also requires cobalamin and iron-sulfur cluster cofactors for turnover. In this work we report the overproduction and purification of TsrM and the characterization of its metallocofactors by UV-visible, electron paramagnetic resonance, hyperfine sublevel correlation (HYSCORE), and Mössbauer spectroscopies as well as protein-film electrochemistry (PFE). The enzyme contains 1 equiv of its cobalamin cofactor in its as-isolated state and can be reconstituted with iron and sulfide to contain one [4Fe-4S] cluster with a site-differentiated Fe(2+)/Fe(3+) pair. Our spectroscopic studies suggest that TsrM binds cobalamin in an uncharacteristic five-coordinate base-off/His-off conformation, whereby the dimethylbenzimidazole group is replaced by a non-nitrogenous ligand, which is likely a water molecule. Electrochemical analysis of the protein by PFE indicates a one-electron redox feature with a midpoint potential of -550 mV, which is assigned to a [4Fe-4S](2+)/[4Fe-4S](+) redox couple. Analysis of TsrM by Mössbauer and HYSCORE spectroscopies suggests that SAM does not bind to the unique iron site of the cluster in the same manner as in other radical SAM (RS) enzymes, yet its binding still perturbs the electronic configuration of both the Fe/S cluster and the cob(II)alamin cofactors. These biophysical studies suggest that TsrM is an atypical RS enzyme, consistent with its reported inability to catalyze formation of a 5'-deoxyadenosyl 5'-radical.
J Am Chem Soc
2016;138(10):3416-3426
| PubMed ID:
26841310
Benjdia A, Pierre S, Gherasim C, Guillot A, Carmona M, Amara P, Banerjee R, Berteau O
The thiostrepton A tryptophan methyltransferase TsrM catalyses a cob(II)alamin-dependent methyl transfer reaction
▸ Abstract
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a novel class of natural products including several antibiotics and bacterial toxins. In countless RiPP biosynthetic pathways, cobalamin-dependent radical SAM (B12/rSAM) enzymes play a pivotal role. In the biosynthetic pathway of the antibiotic and anti-cancer agent thiostrepton A, TsrM, a B12/rSAM enzyme, catalyses the transfer of a methyl group to an electrophilic carbon atom of tryptophan. Here we show that methylcob(III)alamin is the probable physiological enzyme cofactor, and cob(II)alamin rather than cob(I)alamin is a key reaction intermediate. Furthermore, we establish that TsrM and a triple-alanine mutant alkylate cob(II)alamin efficiently leading to the synthesis of MeCbl. Exploiting TsrM substrate ambiguity, we demonstrate that TsrM does not catalyse substrate H-atom abstraction like most radical SAM enzymes. Based on these data, we propose an unprecedented radical-based C-methylation mechanism, which further expands the chemical versatility of rSAM enzymes.
Nat Commun
2015;6(None):837-8377
| PubMed ID:
26456915